Navigator Alpha: Run Your Own Genomics Lab on Your Own Machine
Navigator Alpha
For a while now I have been writing about the pieces: rewriting the stack in Rust, building a Y and mt tree from scratch, what a pangenome catches and misses, and the Folding@Home-style compute grid that could reprocess public genomes onto a modern reference. Those posts described a plan. This one is about the thing you can actually download and run.
Navigator is the desktop application at the center of Decoding Us. It is entering an Alpha test, and this post is the invitation. This post describes what works today and what is still on the roadmap, because an Alpha is exactly the moment to be clear about that.
Where this came from
If you followed the older project, you knew YDNA-Warehouse.org. It let people contribute Y sequencing results and basic genealogy to build a human-scale family tree, and it fed data to other community sites for years. It also closed in May 2025, and the reason it closed is the whole point of what came next.
The Warehouse ran the way nearly every genomics service runs: you uploaded your BAM file, it landed in the cloud, and a server farm did the standardizing and the matching. That model has two costs that never go away. First, compute: every sample has to be aligned and called and placed, and doing that on rented cloud machines costs real money for every genome. Second, storage: a single whole-genome BAM is tens of gigabytes, and once thousands of people have uploaded theirs, you are paying month after month to hold multiple terabytes across several storage tiers. Those two line items are what a large hobby project cannot outrun. Eventually the bill wins.
Decoding Us answers both by moving the work to the edge, to your own computer. Your BAM never leaves your machine, so there is no upload and no terabytes to warehouse. The analysis runs on hardware you already own and already paid for, so there is no per-sample cloud compute bill. What travels the network is only the small result (a haplogroup placement, an ancestry summary, a set of variant observations), and only if you choose to share it. The heavy, expensive parts stay local. The shared network only ever handles the lightweight conclusions. That is the entire architectural bet, and Navigator is where it lives.
What Navigator actually does
Point it at a file and it figures out the rest. It reads BAM and CRAM alignments directly; it also imports VCF/gVCF, consumer chip raw data (23andMe, AncestryDNA), Y-SNP and STR exports, mitochondrial FASTA, and Complete Genomics masterVar files. A header probe infers the reference build, aligner, and platform, then it runs the analysis: coverage and callable regions, read metrics, sex, structural variants, Y and mitochondrial haplogroup placement against our tree, and an ancestry profile with admixture and PCA. Results are saved to a local workspace and can be exported as reports.
The important part is what it is not: there is no JVM, no GATK, no samtools, no bcftools, and nothing to download and configure alongside it. It is a single self-contained program, written in Rust on the noodles bioinformatics stack, that carries its own analysis engine. You install one thing and you are done.
One deliberate boundary is worth stating plainly. The callers are built for genealogy and anthropology, and they are tuned to that standard: placing you on the haplogroup tree, estimating your ancestry, comparing your lineage to others and to the public record. That is the level of quality they aim for and reach. Bridging the gap to clinical or health-diagnostic work is an explicit non-goal. Navigator is not a medical device, it is not validated for diagnosis or health discovery, and nothing it reports should be read as clinical information. Health-grade calling carries regulatory and validation burdens that are a different project entirely, and trying to serve both masters would compromise the one this tool is actually for. If you want to know where you sit on the tree and where your ancestors came from, that is exactly what these callers are for. If you want a health interpretation, this is the wrong tool, on purpose.
None of that stops you from bringing higher-grade calls to the table when you have them. If you already run a proven, validated variant-calling workflow of your own, or you have VCFs a clinical pipeline produced, you do not have to re-call anything in Navigator. Import that VCF through the sidecar system instead, and Navigator will use your calls as the input to the genealogy and anthropology work it does downstream. Bring your own trusted variants; let Navigator do the tree placement, ancestry, and consensus on top of them.
Installing it
For most people the intended path is the simplest one: grab a prebuilt installer from GitHub Releases, download, install, and launch. The current Alpha build is v0.1.0-alpha.7, and there is a package for every common desktop:
| Platform | Download |
|---|---|
| macOS (Apple Silicon + Intel, universal) | DUNavigator_0.1.0_universal.dmg |
| Windows (x64) | navigator_0.1.0_x64-setup.exe |
Linux .deb (x86_64 / amd64) |
navigator_0.1.0_amd64.deb |
Linux .deb (ARM64) |
navigator_0.1.0_arm64.deb |
| Linux AppImage (x86_64) | navigator_0.1.0_x86_64.AppImage |
| Linux AppImage (ARM64 / aarch64) | navigator_0.1.0_aarch64.AppImage |
The macOS bundle is a signed, notarized universal .dmg that runs natively on both Apple Silicon and Intel, and it has been built and validated end to end, so that is the front door I expect most macOS testers to use. On Linux, take the .deb if you are on a Debian/Ubuntu-family distribution and the AppImage if you would rather have a single self-contained executable. Each release also ships a SHA256SUMS file if you want to verify your download. Because these are Alpha builds, newer tags will land as bugs get fixed; the latest release page always points at the freshest packages.
Because the whole thing is one Rust binary with no external tools, building from source is genuinely easy, and that is the path if you are on a platform I do not ship a prebuilt installer for. FreeBSD is the obvious one, but the same applies to less common Linux setups or anyone who simply prefers to build their own:
git clone https://github.com/JamesKane/decodingus-navigator
cd decodingus-navigator
cargo build --release
If you have a working Rust toolchain, that is the whole recipe; the resulting binary is named navigator. There is no pipeline to assemble, no reference bundle to hunt down by hand. Navigator fetches and caches the reference genomes it needs on first use. On FreeBSD, the Rust toolchain is a pkg install away, so “install Rust, cargo build --release” really is the whole story.
Run the binary with no arguments and you get the desktop app. Run it with a subcommand (ingest, subjects, show, projects) and you get a headless CLI over the same engine and the same workspace, handy for a home server or for scripting a batch of files.
The first run: one sample, done for you
The default experience is deliberately boring in the best way. You have one DNA file. You bring it in, Navigator detects what it is, and runs only the analysis that file can actually support. What you see is scoped to what your test measured. A whole-genome BAM has the coverage for the full picture: Y haplogroup and the path to it, mitochondrial haplogroup, coverage, an ancestry breakdown. A targeted test gives you the part it covers and nothing it cannot honestly produce. A FTDNA Big Y is a Y-only test, so it gets a Y haplogroup placement and Y coverage, and Navigator does not invent an mtDNA or ancestry result it has no data for. A mitochondrial FASTA gives you your mtDNA haplogroup and its mutations. A consumer chip export gives you the ancestry and the low-resolution haplogroup calls the chip can support. You do not have to know what a callable region is or which reference build your test used, and you are never shown a result the file could not back up. That is the single-sample path, and for most people it is the entire app. Bring a file, read your results.
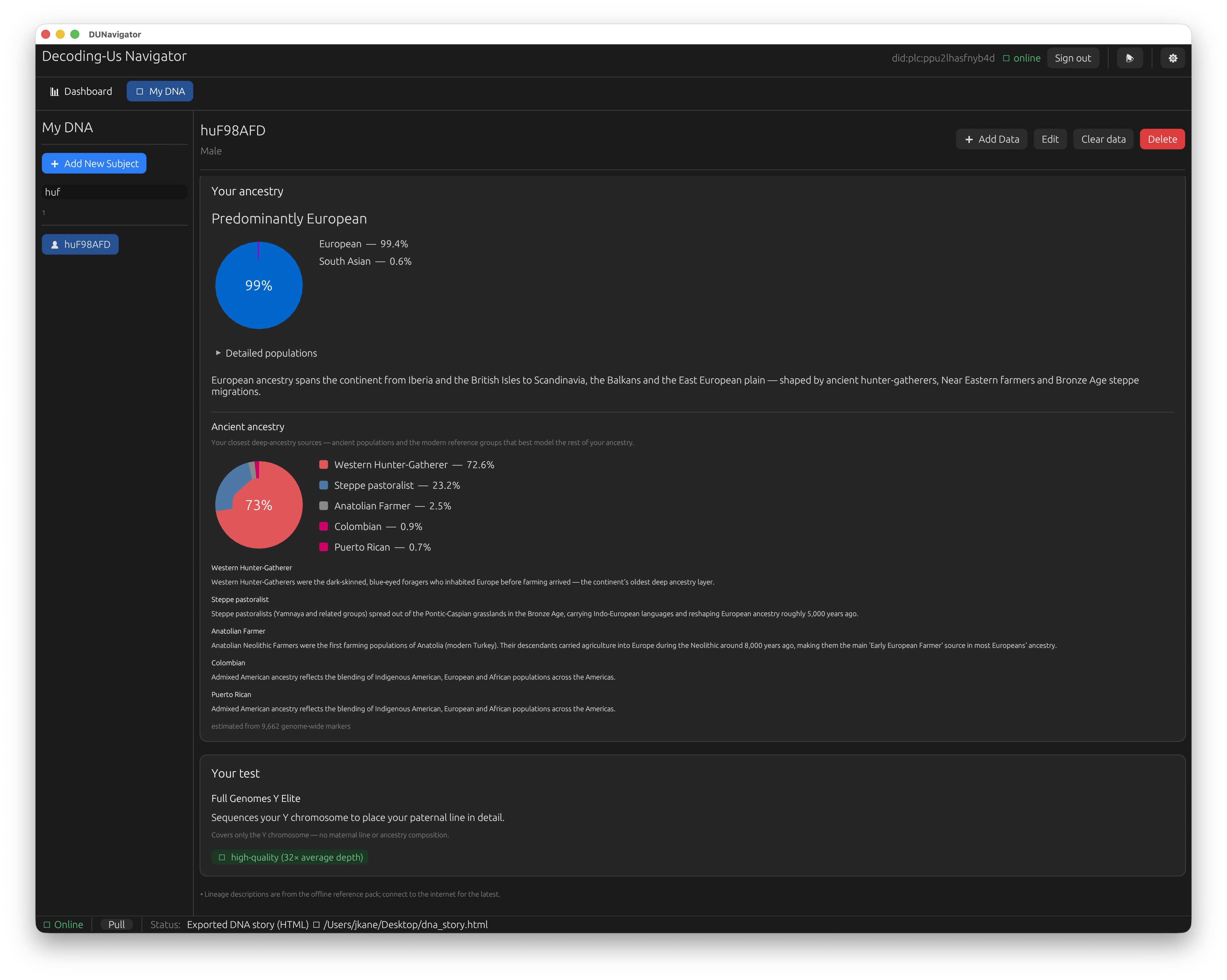
The simplified “My DNA” view from one whole-genome import: your ancestry composition, the ancient populations that best model it, and a plain-language read on the test itself — no jargon, no knobs to set.
If you want the full walkthrough, the Navigator User Guide covers every feature in depth: importing each supported file type, the project-import batch and sidecar fast path, what each analysis produces, exporting reports, the CLI, and bringing your own reference genomes. This post is the tour; the guide is the manual.
One person, many tests: the progressive consensus genome
Most people in this hobby did not take one test. Over the years they accumulated a few: a Y test from one vendor, a mitochondrial test from another, a 23andMe or AncestryDNA chip from a third. Each one measures a different slice of you, and on its own each is a partial answer.
Navigator lets you attach all of them to the same subject and builds a progressive consensus from them: a whole-genome-like profile assembled out of the pieces. Your Y placement comes from the Y test, your maternal line from the mitochondrial test, your ancestry from the chip, and together they add up to a fuller portrait than any single kit could give. As you add tests, the profile fills in. Feed it a later whole-genome BAM and the same consensus absorbs it, upgrading the pieces that were previously low-resolution.
The valuable part is the cross-checking between technologies. When two tests overlap, Navigator compares them instead of blindly trusting the newest one. A chip’s low-resolution Y call and a Big Y’s deep placement should agree on the branch even if the Big Y goes much deeper; a mitochondrial haplogroup from a targeted test and from a whole-genome BAM should land on the same node. When they agree, that is real confidence that the result is right. When they disagree, that is worth knowing too, because it points at a genotyping error, a sample mix-up, or a mislabeled file, and Navigator surfaces the conflict rather than quietly papering over it. Consensus across independent measurements is a stronger claim than any one measurement alone.
Underneath that simple surface there is an advanced side for people who want it. Navigator’s workspace holds multiple subjects, and you can group them into projects, the way a surname or haplogroup project organizes many kits, with overview charts and side-by-side comparison across the members. If you administer a project, or you are working through a family’s worth of kits, that structure is there. If you have one kit and one question, you will never need to touch it. The design goal is that the simple case stays simple and the powerful case is one layer down, not in your face.
The optional AI helper
Navigator can talk to a local large language model (LM Studio or Ollama running on your own machine) to narrate your results, answer plain-language questions about them, and explain what a given tab is showing. It is aimed squarely at newcomers who would rather ask “what does this ancestry chart mean?” than read documentation.
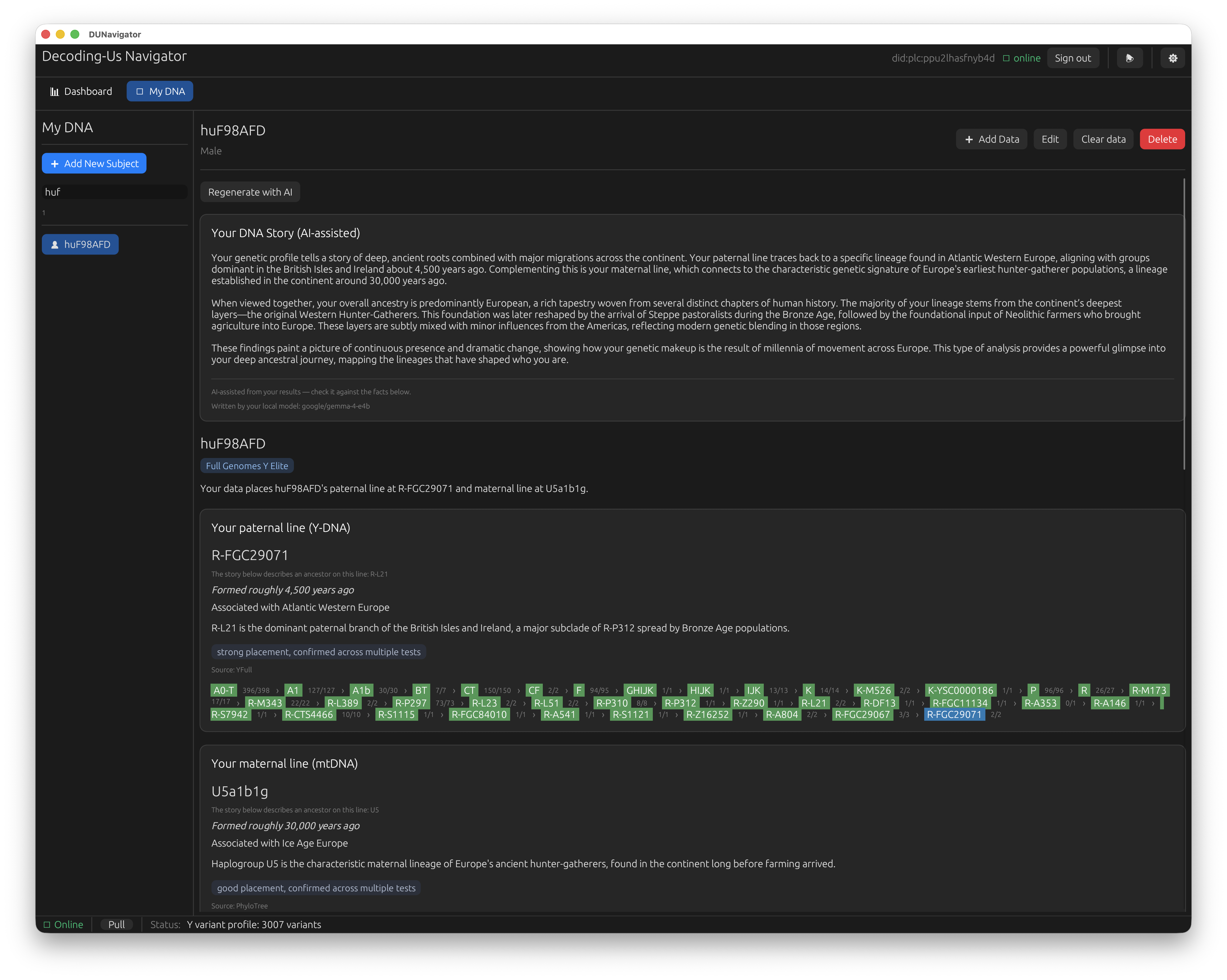
The same results, narrated. A local model — here google/gemma-4-e4b — writes the “DNA Story” from your computed placements, right above the lineage cards it is summarizing. The “written by your local model” line and the facts printed just below it keep the generated prose honest and grounded.
Two things to be clear about. It is entirely optional: nothing in the analysis depends on it, and if you never set it up you lose no capability, only the conversational help. And it is local: the model runs on your computer, so your results are not being shipped to some external AI service to be explained. If you have no interest in it, ignore it and Navigator works exactly the same.
Contributing to the tree
Everything above works fully offline and on its own. But the reason Decoding Us exists is the shared, community-built haplogroup tree, and that only grows if people contribute their placements and observations back. That part is opt-in, and it runs on the AT Protocol, the same federated network behind Bluesky.
To contribute, you sign in with AT Protocol credentials, and Navigator publishes your results (placements and variant observations, not your raw genome) to your own data store on the network. A couple of recommendations for how to do that comfortably:
- Use a dedicated profile, not your main Bluesky account. Make a separate handle for your genomics contributions and sign Navigator in with that. It keeps your genealogy activity cleanly separated from your personal social account, and if you ever want to hand off or retire the contributing identity, you can do it without touching your everyday presence.
- A private PDS is nice to have, not required. In AT Protocol terms your data lives in a Personal Data Store. Running your own PDS gives you the fullest ownership, but self-hosting one is genuinely a homelab project, and I am not going to pretend otherwise. If you are not the sort of person who enjoys standing up servers, use a hosted PDS (the default Bluesky one is fine) and you still keep control of your records and can move them later. Self-hosting is the enthusiast option, not the price of admission.
If you never sign in at all, Navigator is still a complete local analysis tool. Contributing is the part where your results join everyone else’s and the tree gets denser, but it is a choice, not a toggle you have to flip to get value.
What this Alpha is for
The current focus is fixing bugs and polishing the experience. The analysis engine is real and the results are ones I trust. This is the same stack I have been validating against my own kit and a corpus of others across these posts. What I need from an Alpha is people running it on hardware I have never seen, on files from tests I have not personally handled, hitting the rough edges I cannot find alone. Rough edges are the expected output. Reporting them is the contribution.
The bigger systems I have written about are deliberately not in this release. The re-alignment engine that reprocesses old alignments onto the modern T2T reference, and the SETI@home-style compute grid that would let volunteers reprocess public genomes overnight: those are specified and their foundations are built, but they come in later releases. This Alpha is about getting the thing in your hands solid first: one person, one machine, one file, results you can trust. The distributed future is more fun to write about, but it is worth nothing if the everyday experience is not dependable, and dependable is what this phase is for.
If you want to be part of that, grab the installer, point it at a file, and tell me what breaks.
For technical details, visit decoding-us.com, read the User Guide, or check out the source code.